This web page was produced as an assignment for Genetics 564, an undergraduate capstone course at UW-Madison.
Phylogeny
Phylogeny is the study of evolutionary relationships between species (1). These relationships can be studied using a variety of statistical methods, the most common methods being maximum likelihood and neighbor-joining. Phylogeny can be used to identify the evolutionary relationships between species, but also between specific protein sequences.
Neighbor-joining uses percent identity/BLOSSUM similarity scores to determine which species are most closely related, and uses physical distance to represent evolutionary difference (2).
Maximum likelihood uses a previously made tree by another method, then calculates the probability of the tree and adjusts branch length accordingly (2).
Average distance takes scores generated by sequence similarities and joins the identical species together with branches that are equal in length (2).
Maximum likelihood uses a previously made tree by another method, then calculates the probability of the tree and adjusts branch length accordingly (2).
Average distance takes scores generated by sequence similarities and joins the identical species together with branches that are equal in length (2).
Constructing a Phylogenetic Tree for PINK1
1. Obtain Homolog Sequences in FASTA format (save as .txt file)
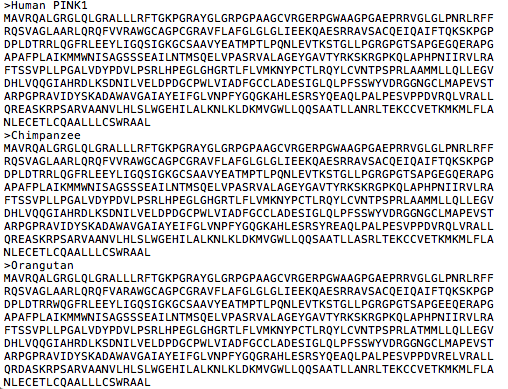
2. Align sequences with ClustalOmega
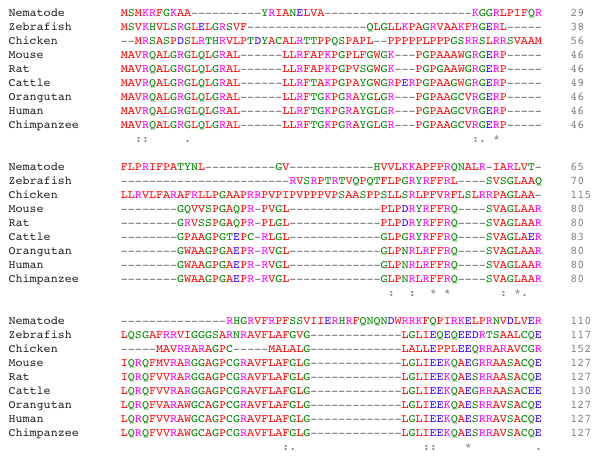
3. Create simple tree using ClustalOmega
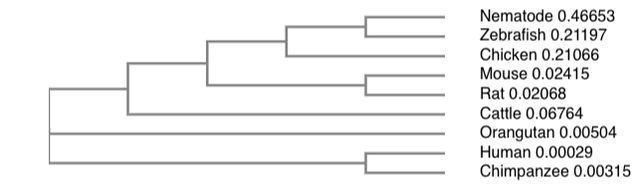
4. Create trees using MEGA
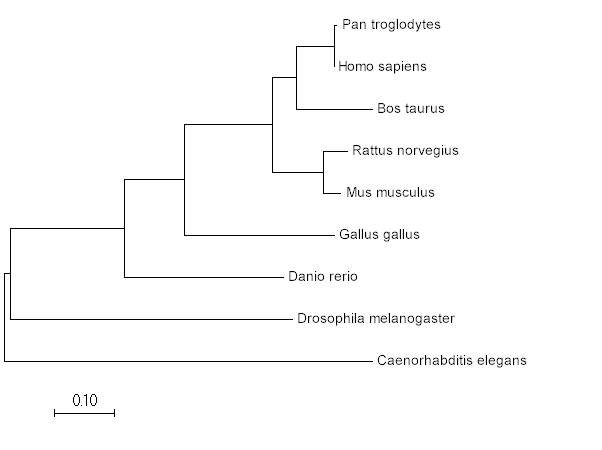
Maximum Likelihood
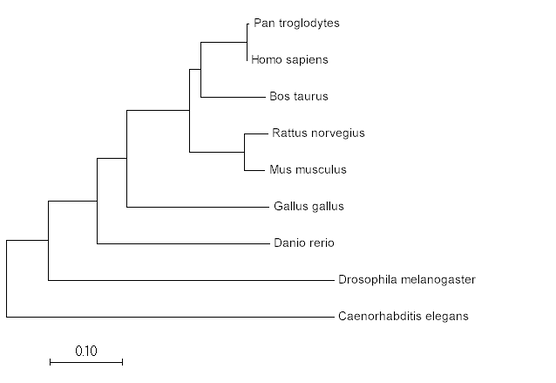
Neighbor Joining
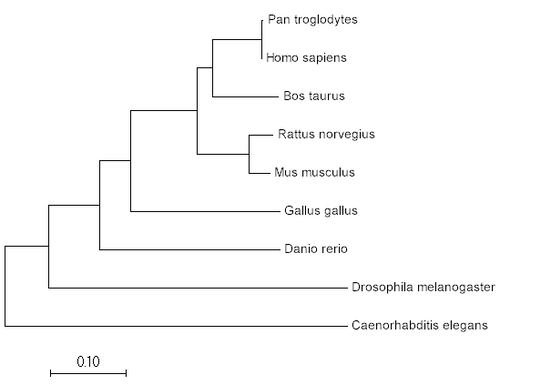
Minimum Evolution
1. University of California-Berkeley. (2008). "Phylogenetics." Retrieved from http://evolution.berkely.edu/evolibrary/article/phylogenetics_02
2. http://evolution-textbook.org/content/free/contents/ch27.html#ch27-4-2
2. http://evolution-textbook.org/content/free/contents/ch27.html#ch27-4-2
